Mapování QTL
Živočišný genom
Genom je definován jako celková suma genetické informace v konkrétní buňce nebo organizmu. Konkrétnější definice říká, že genom jsou všechny geny nesené jednou gametou, tedy haploidní sadou chromozomů. Rozlišujeme genom jaderný a mimojaderný (mitochondriální). Mitochondriální DNA je součástí genomu. Vědní obor zabývající se studiem genomu se nazývá genomika, která má dva primární cíle: určit sekvence každého chromozomu v genomu a identifikovat polymorfizmy nebo alelickém varianty přítomné v genomu. Tato genomika se také nazývá strukturní genomika. V postsekvenačním období současnosti nabývá na významu studium vztahů mezi geny v genomu a mezi geny a proteiny. Touto částí genomiky se zabývá funkční genomika.
Genomy žijících organizmů jsou významně odlišné ve své velikosti. Genomy mikroorganizmů a eukaryotů dosahují rozpětí od 700 000 bp (700 kb) u jednobuněčného mikrobiálního chromozomu až po více než 3 mld. bp (3 Gb), v rozmezí od 5 do 96 chromozomů u různých savců. Haploidní genom člověka obsahuje přibližně 3 miliardy nukleotidů DNA rozmístěných na 23 chromozomech, v kterých je asi 25 000 genů. Podobnou velikost genomu mají i významné druhy hospodářských zvířat.
Projekt mapování lidského genomu je spojen s rozvojem nových technologií, studiem modelových organizmů, analýzou variability genomu a vývojem bioinformatiky. DNA sekvencování je proces určující specifické pořadí a identifikaci všech nukleotidů (párů bazí) v genomu, aby mohly být identifikovány jednotlivé geny. Mapování je proces identifikace samostatných molekul DNA se známou pozicí na chromozomu, které jsou pak používané k sekvencování. Mapování je rozhodující krok pro správnou rekonstrukci genomu. Obvykle předchází sekvencování, ale je také nutné v postsekvenačním období současnosti.
Genetické mapy a mapování
Pod pojmem genetická mapa rozumíme znázornění distribuce určitého počtu genů v genomu. Mapování může být prováděno různými metodickými postupy a podle toho rozeznáváme mapy:
- Cytogenetické (chromozomové) mapy – založeny na fyzické podobě chromozomů (karyotyp). Za účelem identifikace je možno určitým způsobem upravené a obarvené chromozomy pruhovat a porovnávat se standardními ideogramy. Poloha genu na cytogenetické mapě je udávána číslem chromozomu, písmenem označujícím raménko a čísly jednotlivých oblastí dle pruhů. U druhů, které mají dobře definovaný karyotyp (např. prase), se ke konstrukci cytogenetických map používá hybridizace in situ. Lokalizace genu je prováděna pomocí hybridizace určitého značeného úseku DNA (sonda) s denaturovanou DNA chromozomu. Značení se provádí radioaktivně a dnes převážně fluorescenčně – FISH. Metoda je založena na fůzi buněk dvou biologických druhů (např. skot a křeček), čímž vznikne buněčná linie, která postupně ztrácí chromozomy a v konečné fázi je stabilizována s jedním nebo několika chromozomy vyšetřovaného druhu (skot) a kompletní chromozomovou sadou druhého druhu (křeček). Podobné metody se používají i při mapování QTL.
- Fyzické mapy – jsou založeny na přímé analýze DNA. Vzdálenosti mezi jednotlivými lokusy se určují počtem párů bazí.
- Vazbové (rekombinační) mapy – informují nás o vzdálenosti a pořadí genů na chromozomu. Vazba je detekovaná na základě počtu crossing-overů a udává se v jednotkách procentech rekombinací (1 % = 1 cM - centi morgan). Vazba mezi geny existuje tehdy, je-li rekombinační frekvence menší než 0,5. Ke statistickému hodnocení průkaznosti a těsnosti vazby je používán test zvaný LOD score. Jedná se o logaritmus poměru pravděpodobnosti (z) existence (0 – 0,5) či neexistence (0,5) vazby. Je-li z > 3 je považována vazba za prokázanou. Je-li menší než 2 je vazba vyloučena. Analýza vazby je prováděna pomocí počítačových programů. Jako velmi výhodné pro vazbové mapování se považují vysoce polymorfní mikrosatelity, které jsou však anonymní a nejsou použitelné pro komparativní mapování. K vyhledávání alelických variant se u skotu používají rodiny polosourozenců a u prasat tří generační rodokmeny vzniklé křížením kontrastních plemen či linií. Analýza vazby je hlavním principem při mapování QTL.
- Komparativní (srovnávací) mapy – jedná se o mapy, na kterých jsou vyznačeny homologní úseky s geny u různých druhů. Takto lze vyhledávat kandidátní geny pro QTL.
Význam a mapování QTL u hospodářských zvířat
Odhad plemenných hodnot je v současné době prováděn analýzou fenotypu nebo fenotypové užitkovosti zvířat. Pokročilé statistické metody (BLUP, AM) se využívají pro oddělení genetických a prostřeďových efektů, při spojování zvířat genetickými příbuzenskými vztahy (Henderson, 1984). Fenotyp však nemůže být vždy zaznamenatelný z důvodů fyziologických (např. býci neprodukují mléko, kanci nemají selata, krávy rodí telata až po dvou letech života, …), některé vlastnosti nelze zjistit u živých zvířat (např. ukazatele jatečního těla, kvalita masa, …) nebo zaznamenávání fenotypu je chybné nebo nepřesné (snadnost telení). V posledních desetiletích došlo k hlubšímu poznání struktury DNA a působení určitých genů. Tyto výzkumy umožňují zkoumat, zda variabilita v DNA (polymorfizmus) určitých zvířat může být vázána k diferencím v produkčních nebo jiných ekonomických vlastností. Je zde však několik zásadních problémů vlastní celému genomu. Musí se určit funkční struktura chromatinu a pak nalézt v této struktuře funkční oblasti (geny) a nefunkční oblasti.
V mapování genomu se největší pozornost soustřeďuje na ty QTL, které by mohly být využity při šlechtění, vedoucí ke zlepšení ekonomiky produkce. Tato QTL se nazývají lokusy ekonomicky významných znaků a označují se ETL. QTL mají vesměs polygenní charakter s bezpočtem kombinací alel a berou se jako celek a o jejich složení se prakticky nic neví. Proto se jejich vztah k užitkovosti vyjadřuje nepřímo prostřednictvím genetických markerů – polymorfních úseků DNA s rozličnou funkcí či bez funkce, ale se známou identifikovatelnou pozicí. V případě, že je polymorfní lokus asociován s užitkovou vlastností, jedná se pravděpodobně o důsledek vazby s QTL. Proto mapování QTL vyžaduje populaci zvířat, ve které je genetická proměnlivost ve zkoumaných vlastnostech a genotypování těchto populací pro panel polymorfních genetických markerů pokrývajících genom v určité hustotě.
Hlavní výhody QTL/ETL ve šlechtitelských programech jsou:
- zvýšení přesnosti v selekci pomocí doplňkových informací přímo vztažených ke genotypu jedince,
- možnost snížit generační interval přidáním nového selekčního období v životě zvířete – nejranější věk, protože QTL/ETL dovolují získat informace nezávislé na pohlaví a věku. Může také dojít ke zvýšení účinnosti introgrese nebo předpovídat heterozu pomocí genetické distance.
Předpoklady dobrého mapování:
- Geny kontrolující kvantitativní vlastnosti mohou být mapovány v genomu jako jednoduché genetické markery.
- Jestliže markery pokrývají větší část genomu, pak je velká šance, že některé geny kontrolující kvantitativní vlastnosti budou vázané k určitým genetickým markerům. Jestliže geny (QTL) a markery segregují v geneticky definované populaci, pak vazbové vztahy mezi nimi mohou být odhalitelné při hledání asociací mezi variabilitou vlastnosti a segregujícím markerem.
- Jestliže genotypy genů kontrolujících kvantitativní vlastnosti mohou být zaznamenány, pak se problémem stává jednoduchá vazbová analýza. Ve skutečnosti nemohou být geny pozorovatelné a to co pozorujeme jsou hodnoty kontinuální vlastnosti.
Detekce QTL/ETL
Tento krok tvoří studium potenciální vazby mezi polymorfizmy DNA a fenotypovými vlastnostmi. Hlavní ideou je, že specifický gen nazývaný QTL/ETL je zodpovědný za část variability daného fenotypu. K detekci QTL/ETL se používají dva přístupy. Přístup kandidátních genů využívá znalost již známých genů, nazývaných jako kandidátní pro QTL/ETL. Je-li znám jen polymorfizmus DNA, pak se předpokládá že tento marker pro danou oblast DNA se nachází blízko QTL/ETL – přístup genetických markerů (Georges et al., 1995).
Metoda kandidátních genů závisí na a priory předpokladu, že známe působení kandidátního genu na genetické a fyziologické úrovni. Musí být známy geny a jejich sekvence DNA. Výběr správného genu by měl být prováděn na základě dobré znalosti jeho pozice v metabolických drahách. Aditivní efekty pro různé alely těchto kandidátních genů se pak porovnávají navzájem, aby se prokázaly substituční efekty alely. Používají se rozdílné techniky na úrovni populací nebo rodin.
Genetické markery jsou také založeny na známem polymorfizmu DNA, které jsou však většinou v nefunkčních oblastech nebo nejdou funkční jako kandidátní geny. Nejvíce jsou využívány mikrosatelity, oblasti s vysokým polymorfizmem. Nověji se začínají prosazovat jednonukleotidové polymorfizmy (SNP). Pokud takové markery segregují v generacích, mohou být analyzovány jejich vztahy s fenotypovými rozdíly. Tato metoda detekce QTL/ETL je založena na analýze rodin.
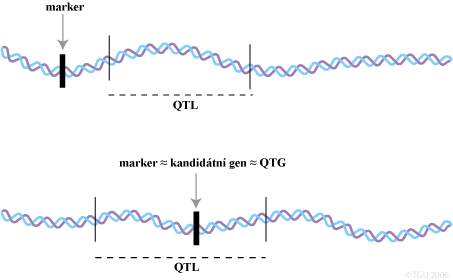
Identifikace mutací v genech zodpovědných za vlastnost nebo v lokusu markeru pro takové geny poskytují genetikům možnost implementovat metodu selekce s podporou markerů (Marker Assisted Selection - MAS). Hlavní výhodou využití genetického kritéria pro selekci je, že taková data mohou být získána v časném věku zvířete a u obou pohlaví. Pokud lokus vykazuje převládající působení a vysvětluje větší část variance produkční vlastnosti je nazýván jako lokus kvantitativních vlastností (QTL). V současnosti se využívají dva způsoby identifikace QTL: metoda kandidátních genů a poziční klonování.
Metoda kandidátních genů
Tato strategie sestává ze studia různých genů, kteří jsou potenciální pro ovlivňování fyziologických procesů (např. syntéza mléčných proteinů) a pro každý gen určení alely, která má za následek žádoucí fenotyp (zlepšené sýřitelnost – např. polymorfizmus k-kaseinu). To vyžaduje znalost genetické kontroly studovaného fyziologického procesu (např. roli somatotropní osy v regulaci laktace). Identifikace polymorfních míst a vyhodnocení rozdílů alel kandidátních genů mezi zvířaty různých fenotypových hodnot nabízí potenciální možnost identifikace markerového genu asociovaného s fenotypovou hodnotou.
Metoda pozičního klonování (detekce QTL v genetických markerech)
Detekce v genetických markerech, chromozomálních segmentech zodpovědných za část genetické variability vlastnosti je zcela odlišná metoda. Po meióze obsahují gamety haploidní počet chromozomů v náhodné kombinaci velkých segmentů v každém rodičovském chromozomovém páru. Dvě DNA sekvence (dva lokusy) lokalizované blízko sebe na stejné rodičovském chromozomu jsou s velkou pravděpodobností přenášeny společně do gamet a pak potomkům. Máme-li dva lokusy M a Q a dvojité heterozygoty MQ/mq. Tento zápis znamená, že na jednom chromozomu jsou alely M a Q (například otcovský) a na druhém m a q (mateřský). Počet rodičovských potomků, tj. těch co dostali rodičovský chromozomální segemnt (MQ nebo mq), je větší než počet rekombinovaných potomků, tj. těch co získali rekombinované chromozomální úseky (Mq nebo mQ) v důsledku crosing-overu. Jsou-li rekombinace vzácné, jsou-li lokusy velmi blízce lokalizované, jsou alely M a m většinou asociovány s Q a q v potomstvu po rodičích (jsou ve vazbové nerovnováze). Jestliže alela Q ovlivňuje vlastnost, pak by mohl být efekt substituce q alelou Q detekován porovnáním dvou skupin potomků mající M a m alely. Ačkoliv marker M geneticky nedeterminuje vlastnost, může detekovat QTL právě pomocí genetické vazby.
Účinnost této metody primárně závisí na charakteristice markerů:
- QTL mohou být lokalizován kdekoliv, také markery mohou být po celém genomu,
- aby mohl být charakterizován a vyhledáván každý rodičovský chromozom, marker by měl být polymorfní, tzn. měl by mít nejméně 2 alely,
- mělo by být možné genotypování markeru, jednouše a levně.
Tyto omezení vysvětlují, proč tato metoda popsaná už v 60. letech byla aplikována až nyní. Dva základní objevy otevřely cestu detekce QTL: objevení polymorfních míst po celém genomu a objev polymerázové řetězové reakce (PCR) pro odhalení těchto polymorfizmů. V posledním desetiletí dochází ke konstrukci čím dál více hustších genetických map hospodářských zvířat.
Strategii využití asociace marker-QTL k lokalizaci QTL lze rozdělit do těchto kroků:
- identifikace chromozomální oblasti obsahující studovaný QTL (10 – 20 cM),
- specifikace lokalizace QTL v této oblasti (5 cM),
- identifikace markerů v těsné vazbě s QTL (1 – 2 cM),
- identifikace potenciálního kandidátního genu v této oblasti.
Tato metoda využívá zejména hustou síť mikrosatelitů a nověji i SNP, dokud není vhodný kandidátní gen identifikován. Proto se tato metoda nazývá poziční klonování.
Vybrané geny
Genetické markery identifikované u skotu (výběr)
- laktace
kandidátní geny
- růstový hormon a zvýšení produkce mléka: somatotropní osa v regulaci laktaci
- GHRH (somatocrinin, growth hormone releasing hormone; chr. 13) a GHRHR (receptor; chr. 4) – regulují GH
- GH (růstový hormon; chr. 19) a GHR (receptor; chr. 20)
- buněčně specifický transkripční faktor pro aktivaci prolaktinu (PRL) a GH genů v adenohypofýze
- Pit-1 (Pituitary-Specific Transcription Factor) (chr. 1)
- IGF-1 (Insuline growth factor 1; chr. 5) a IGF-1 receptor (chr. 21)
poziční klonování
- QTL na chromozomu 1, 2, 3, 5, 6, 7, 8, 9, 10, 14, 16, 18, 20, 23
- produkce masa a kvalita
- kandidátní gen GH
- poziční klonování
- gen svalové hypertrofie - myostatin
- chromozom 5, 6, 14, 17, 27, 29
- IGF-1
Genetické markery identifikované u prasat (výběr)
- GH (chr. 12) (bez asociací)
- Pit-1 (POU domain, class 1, transcription factor 1; chr. 13)
- PRLR (receptor prolaktinu; chr. 16)
- RYR1 (Ryanodine receptor 1; chr. 6)
- RN (Rendement Napole; chr. 15)
- ESR (Estrogen receptor; chr. 1)
Genetické markery identifikované u kura
- GH (chr. 1), GHR (chr. Z)
- NRAMP1 (Natural resistance-associated macrophage protein 1; chr. 7)
- salmonela s chr. 5 – geny rezistence
U hospodářských zvířat již bylo zmapováno velké množství QTL. Mohou se stát silným nástrojem k tvorbě ideálního genotypu pro daná prostředí. Mnoho autorů zdůrazňuje, že je nutné potvrdit přítomnost zmapovaného QTL než se přikročí k dalšímu kroku v MAS. Je třeba testovat mnoho jedinců u různých plemen. Je nutné vyvinout statistické programy, které umožní přesné vyhodnocení vztahu mezi vlastností a variabilitou genu. V současné době se provádí řada simulačních studií, aby se odhadla odpověď na MAS. Biologické systémy jsou komplexní a je nutné počítat i s interakcemi mezi geny a geny a prostředím.
Aktualizováno: 09.12.2008